Theme: Calibration, plausibility, and responsible reporting
Why This Lesson Exists
A statistically significant association is not automatically a biological discovery.
GWAS identifies statistical relationships between genotype and phenotype. It does not automatically establish causality, mechanism, or clinical relevance.
This lesson focuses on disciplined interpretation.
Recompute Results (Self-Contained)
source("scripts/R/cdi-plot-theme.R")library(ggplot2)pheno <-read.csv("data/demo-phenotype.csv", stringsAsFactors =FALSE)covar <-read.csv("data/demo-covariates.csv", stringsAsFactors =FALSE)geno <-read.csv("data/demo-genotypes.csv", row.names =1)df <-merge(pheno, covar, by ="sample_id")common_ids <-intersect(df$sample_id, rownames(geno))df <- df[match(common_ids, df$sample_id), ]geno <- geno[common_ids, , drop =FALSE]test_snp_lm <-function(x, df){if (any(is.na(x))){ x[is.na(x)] <-mean(x, na.rm =TRUE) } fit <-lm(trait ~ x + age + sex + PC1 + PC2, data = df) co <-summary(fit)$coefficientsc(beta =unname(co["x", "Estimate"]),se =unname(co["x", "Std. Error"]),p =unname(co["x", "Pr(>|t|)"]) )}res <-t(apply(geno, 2, test_snp_lm, df = df))res <-as.data.frame(res)res$snp <-rownames(res)res$logp <--log10(res$p)alpha_nominal <-0.05res$is_sig <- res$p < alpha_nominalres <- res[order(res$p), ]head(res, 5)
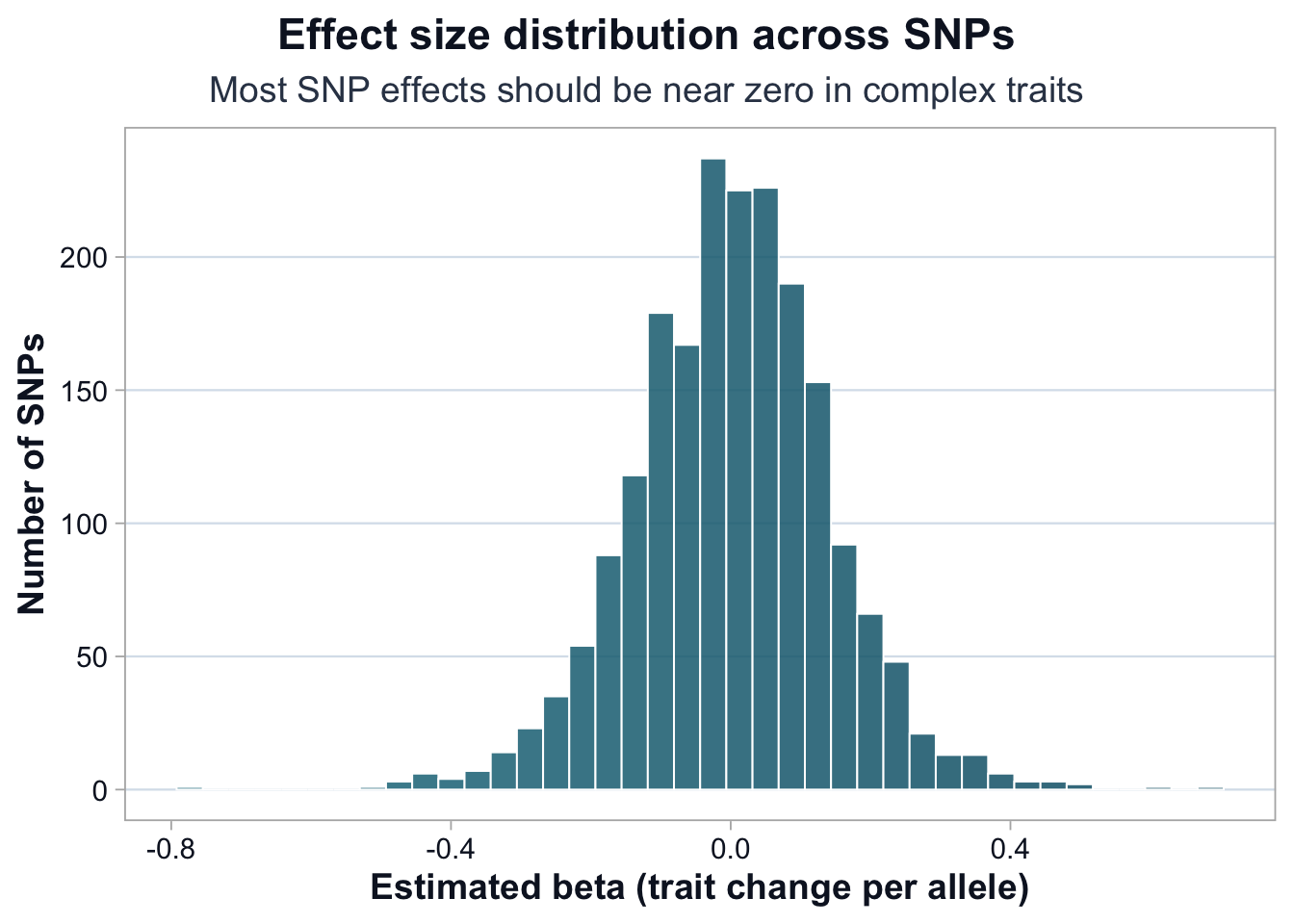
ggplot(res, aes(x = beta)) +cdi_geom_histogram(bins =40, colored =TRUE) +cdi_scale_histogram_fill() +labs(title ="Effect size distribution across SNPs",subtitle ="Most SNP effects should be near zero in complex traits",x ="Estimated beta (trait change per allele)",y ="Number of SNPs" ) +cdi_theme()
Interpretation
In complex traits, most variants have small effects. A symmetric distribution centered near zero is expected under polygenicity. Large-magnitude effects should be treated with caution and verified carefully.